

What Atom Helps Makeup Amino Acids, The Disulfide Bonds Help Add Structure To Big Proteins
From a chemic point of view, proteins are by far the most structurally circuitous and functionally sophisticated molecules known. This is perhaps not surprising, once one realizes that the structure and chemical science of each protein has been developed and fine-tuned over billions of years of evolutionary history. We start this chapter by because how the location of each amino acid in the long string of amino acids that forms a protein determines its three-dimensional shape. We will then use this understanding of protein structure at the atomic level to depict how the precise shape of each poly peptide molecule determines its office in a cell.
The Shape of a Poly peptide Is Specified by Its Amino Acrid Sequence
Recall from Affiliate 2 that there are 20 types of amino acids in proteins, each with different chemic properties. A protein molecule is made from a long chain of these amino acids, each linked to its neighbour through a covalent peptide bond (Figure 3-1). Proteins are therefore also known equally polypeptides. Each type of protein has a unique sequence of amino acids, exactly the aforementioned from ane molecule to the side by side. Many thousands of dissimilar proteins are known, each with its own particular amino acid sequence.
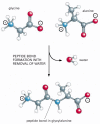
Effigy three-ane
A peptide bond. This covalent bond forms when the carbon atom from the carboxyl group of i amino acid shares electrons with the nitrogen atom (blueish) from the amino group of a second amino acrid. Equally indicated, a molecule of water is lost in this condensation (more than...)
The repeating sequence of atoms forth the core of the polypeptide chain is referred to equally the polypeptide backbone. Attached to this repetitive chain are those portions of the amino acids that are not involved in making a peptide bail and which give each amino acid its unique properties: the 20 different amino acid side bondage (Effigy 3-ii). Some of these side chains are nonpolar and hydrophobic ("water-fearing"), others are negatively or positively charged, some are reactive, and so on. Their atomic structures are presented in Panel 3-1, and a brief list with abbreviations is provided in Figure 3-3.

Figure 3-2
The structural components of a poly peptide. A protein consists of a polypeptide backbone with attached side bondage. Each type of protein differs in its sequence and number of amino acids; therefore, it is the sequence of the chemically different side chains (more than...)
![]()
Panel iii-i
The 20 Amino Acids Establish in Proteins.

Figure iii-3
The 20 amino acids found in proteins. Both three-letter and one-letter of the alphabet abbreviations are listed. As shown, there are equal numbers of polar and nonpolar side bondage. For their atomic structures, see Panel iii-1 (pp. 132–133).
As discussed in Chapter two, atoms carry almost as if they were hard spheres with a definite radius (their van der Waals radius). The requirement that no two atoms overlap limits greatly the possible bond angles in a polypeptide chain (Figure iii-4). This constraint and other steric interactions severely restrict the diversity of three-dimensional arrangements of atoms (or conformations) that are possible. However, a long flexible chain, such as a poly peptide, can still fold in an enormous number of means.

Figure 3-iv
Steric limitations on the bail angles in a polypeptide concatenation. (A) Each amino acid contributes three bonds (blood-red) to the backbone of the chain. The peptide bond is planar (gray shading) and does non permit rotation. Past contrast, rotation tin can occur about (more than...)
The folding of a protein chain is, however, farther constrained past many different sets of weak noncovalent bonds that grade betwixt 1 part of the chain and another. These involve atoms in the polypeptide backbone, every bit well every bit atoms in the amino acid side chains. The weak bonds are of three types: hydrogen bonds, ionic bonds, and van der Waals attractions, as explained in Chapter 2 (run into p. 57). Individual noncovalent bonds are 30–300 times weaker than the typical covalent bonds that create biological molecules. Only many weak bonds can act in parallel to concur two regions of a polypeptide chain tightly together. The stability of each folded shape is therefore adamant by the combined force of large numbers of such noncovalent bonds (Figure iii-5).

Figure 3-v
Three types of noncovalent bonds that help proteins fold. Although a single one of these bonds is quite weak, many of them often form together to create a potent bonding arrangement, equally in the example shown. As in the previous figure, R is used as a general (more...)
A 4th weak force also has a cardinal part in determining the shape of a protein. As described in Chapter two, hydrophobic molecules, including the nonpolar side chains of item amino acids, tend to be forced together in an aqueous environment in club to minimize their disruptive outcome on the hydrogen-bonded network of water molecules (run into p. 58 and Panel 2-2, pp. 112–113). Therefore, an of import gene governing the folding of any protein is the distribution of its polar and nonpolar amino acids. The nonpolar (hydrophobic) side chains in a poly peptide—belonging to such amino acids equally phenylalanine, leucine, valine, and tryptophan—tend to cluster in the interior of the molecule (just as hydrophobic oil droplets coalesce in water to grade one large droplet). This enables them to avoid contact with the water that surrounds them inside a cell. In contrast, polar side chains—such every bit those belonging to arginine, glutamine, and histidine—tend to adjust themselves near the outside of the molecule, where they can form hydrogen bonds with water and with other polar molecules (Figure 3-half dozen). When polar amino acids are cached inside the poly peptide, they are ordinarily hydrogen-bonded to other polar amino acids or to the polypeptide courage (Figure iii-7).
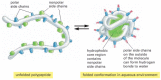
Effigy 3-6
How a protein folds into a meaty conformation. The polar amino acid side chains tend to assemble on the outside of the protein, where they can collaborate with h2o; the nonpolar amino acrid side chains are cached on the inside to class a tightly packed hydrophobic (more...)
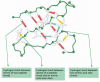
Effigy 3-7
Hydrogen bonds in a protein molecule. Large numbers of hydrogen bonds course betwixt next regions of the folded polypeptide chain and aid stabilize its 3-dimensional shape. The protein depicted is a portion of the enzyme lysozyme, and the hydrogen (more...)
Proteins Fold into a Conformation of Lowest Energy
As a result of all of these interactions, each type of protein has a detail 3-dimensional construction, which is determined by the order of the amino acids in its chain. The terminal folded structure, or conformation, adopted by any polypeptide chain is generally the one in which the gratuitous energy is minimized. Protein folding has been studied in a exam tube by using highly purified proteins. A protein tin be unfolded, or denatured, by treatment with certain solvents, which disrupt the noncovalent interactions belongings the folded concatenation together. This treatment converts the protein into a flexible polypeptide concatenation that has lost its natural shape. When the denaturing solvent is removed, the protein ofttimes refolds spontaneously, or renatures, into its original conformation (Effigy 3-viii), indicating that all the information needed for specifying the 3-dimensional shape of a protein is contained in its amino acid sequence.

Figure iii-viii
The refolding of a denatured protein. (A) This experiment demonstrates that the conformation of a protein is adamant solely by its amino acid sequence. (B) The structure of urea. Urea is very soluble in water and unfolds proteins at high concentrations, (more...)
Each protein unremarkably folds up into a single stable conformation. However, the conformation oftentimes changes slightly when the protein interacts with other molecules in the cell. This modify in shape is oft crucial to the role of the poly peptide, as we see later.
Although a protein chain can fold into its correct conformation without outside help, protein folding in a living jail cell is often assisted past special proteins called molecular chaperones. These proteins bind to partly folded polypeptide chains and help them progress along the nearly energetically favorable folding pathway. Chaperones are vital in the crowded conditions of the cytoplasm, since they prevent the temporarily exposed hydrophobic regions in newly synthesized protein chains from associating with each other to form protein aggregates (see p. 357). However, the final three-dimensional shape of the poly peptide is withal specified by its amino acid sequence: chaperones simply make the folding process more reliable.
Proteins come up in a wide variety of shapes, and they are generally betwixt 50 and 2000 amino acids long. Large proteins generally consist of several distinct protein domains—structural units that fold more than or less independently of each other, equally we hash out below. The detailed structure of any protein is complicated; for simplicity a poly peptide'southward structure tin can exist depicted in several different means, each emphasizing unlike features of the protein.
Panel 3-2 (pp. 138–139) presents four dissimilar depictions of a protein domain called SH2, which has of import functions in eucaryotic cells. Constructed from a string of 100 amino acids, the structure is displayed equally (A) a polypeptide backbone model, (B) a ribbon model, (C) a wire model that includes the amino acid side chains, and (D) a infinite-filling model. Each of the iii horizontal rows shows the protein in a dissimilar orientation, and the image is colored in a mode that allows the polypeptide chain to exist followed from its North-terminus (purple) to its C-terminus (red).
![]()
Panel 3-2
Four different Ways of Depicting a Small Protein Domain: the SH2 Domain (Courtesy of David Lawson.).
Console iii-two shows that a protein'due south conformation is amazingly complex, even for a structure every bit small equally the SH2 domain. But the description of protein structures can be simplified by the recognition that they are built upwardly from several common structural motifs, as we talk over next.
The α Helix and the β Sheet Are Common Folding Patterns
When the three-dimensional structures of many different protein molecules are compared, it becomes clear that, although the overall conformation of each protein is unique, two regular folding patterns are frequently found in parts of them. Both patterns were discovered about 50 years ago from studies of pilus and silk. The offset folding pattern to be discovered, called the α helix, was found in the protein α-keratin, which is abundant in pare and its derivatives—such equally pilus, nails, and horns. Within a year of the discovery of the α helix, a second folded structure, chosen a β sheet, was found in the poly peptide fibroin, the major constituent of silk. These two patterns are particularly mutual because they result from hydrogen-bonding between the Northward–H and C=O groups in the polypeptide backbone, without involving the side chains of the amino acids. Thus, they tin be formed by many dissimilar amino acrid sequences. In each example, the protein chain adopts a regular, repeating conformation. These two conformations, also as the abbreviations that are used to denote them in ribbon models of proteins, are shown in Figure three-9.

Figure 3-nine
The regular conformation of the polypeptide courage observed in the α helix and the β canvas. (A, B, and C) The α helix. The Northward–H of every peptide bond is hydrogen-bonded to the C=O of a neighboring peptide bond located (more...)
The core of many proteins contains extensive regions of β canvass. Every bit shown in Figure 3-x, these β sheets can grade either from neighboring polypeptide chains that run in the aforementioned orientation (parallel bondage) or from a polypeptide chain that folds dorsum and forth upon itself, with each department of the chain running in the direction opposite to that of its immediate neighbors (antiparallel chains). Both types of β sheet produce a very rigid structure, held together by hydrogen bonds that connect the peptide bonds in neighboring chains (see Effigy three-9D).

Effigy 3-10
Ii types of β canvass structures. (A) An antiparallel β sheet (see Figure 3-9D). (B) A parallel β sheet. Both of these structures are common in proteins.
An α helix is generated when a single polypeptide concatenation twists effectually on itself to form a rigid cylinder. A hydrogen bond is made between every fourth peptide bond, linking the C=O of 1 peptide bond to the Northward–H of another (meet Figure 3-9A). This gives rise to a regular helix with a consummate turn every 3.half-dozen amino acids. Note that the protein domain illustrated in Panel 3-two contains two α helices, besides equally β sheet structures.
Brusque regions of α helix are especially arable in proteins located in cell membranes, such equally transport proteins and receptors. As we discuss in Chapter ten, those portions of a transmembrane protein that cantankerous the lipid bilayer usually cross every bit an α helix equanimous largely of amino acids with nonpolar side chains. The polypeptide backbone, which is hydrophilic, is hydrogen-bonded to itself in the α helix and shielded from the hydrophobic lipid environment of the membrane by its protruding nonpolar side chains (come across also Effigy iii-77).
In other proteins, α helices wrap around each other to form a specially stable construction, known as a coiled-curlicue. This construction can course when the two (or in some cases 3) α helices accept most of their nonpolar (hydrophobic) side chains on 1 side, so that they tin twist around each other with these side bondage facing in (Figure 3-11). Long rodlike coiled-coils provide the structural framework for many elongated proteins. Examples are α-keratin, which forms the intracellular fibers that reinforce the outer layer of the skin and its appendages, and the myosin molecules responsible for muscle contraction.
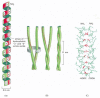
Figure 3-11
The structure of a coiled-whorl. (A) A unmarried α helix, with successive amino acrid side chains labeled in a sevenfold sequence, "abcdefg" (from bottom to top). Amino acids "a" and "d" in such a sequence (more...)
The Protein Domain Is a Fundamental Unit of Organization
Fifty-fifty a small protein molecule is built from thousands of atoms linked together by precisely oriented covalent and noncovalent bonds, and information technology is extremely difficult to visualize such a complicated structure without a three-dimensional display. For this reason, various graphic and computer-based aids are used. A CD-ROM produced to accompany this book contains computer-generated images of selected proteins, designed to exist displayed and rotated on the screen in a variety of formats.
Biologists distinguish four levels of organisation in the construction of a protein. The amino acid sequence is known as the master construction of the poly peptide. Stretches of polypeptide chain that form α helices and β sheets constitute the protein's secondary construction. The full 3-dimensional organisation of a polypeptide chain is sometimes referred to as the poly peptide's tertiary structure, and if a particular protein molecule is formed as a complex of more one polypeptide chain, the complete construction is designated as the fourth structure.
Studies of the conformation, function, and development of proteins take besides revealed the central importance of a unit of organization distinct from the four just described. This is the protein domain, a substructure produced by any part of a polypeptide chain that can fold independently into a meaty, stable structure. A domain unremarkably contains between 40 and 350 amino acids, and information technology is the modular unit from which many larger proteins are constructed. The dissimilar domains of a poly peptide are often associated with different functions. Effigy 3-12 shows an example—the Src protein kinase, which functions in signaling pathways inside vertebrate cells (Src is pronounced "sarc"). This protein has four domains: the SH2 and SH3 domains have regulatory roles, while the two remaining domains are responsible for the kinase catalytic activeness. Afterward in the affiliate, we shall return to this protein, in order to explain how proteins can course molecular switches that transmit information throughout cells.

Effigy 3-12
A protein formed from four domains. In the Src protein shown, ii of the domains form a protein kinase enzyme, while the SH2 and SH3 domains perform regulatory functions. (A) A ribbon model, with ATP substrate in red. (B) A spacing-filling model, with (more...)
The smallest protein molecules contain but a single domain, whereas larger proteins can contain as many every bit several dozen domains, usually connected to each other by curt, relatively unstructured lengths of polypeptide chain. Figure three-thirteen presents ribbon models of three differently organized protein domains. As these examples illustrate, the central core of a domain can exist constructed from α helices, from β sheets, or from various combinations of these two fundamental folding elements. Each different combination is known as a protein fold. So far, virtually 1000 dissimilar poly peptide folds accept been identified amid the ten g proteins whose detailed conformations are known.

Figure 3-thirteen
Ribbon models of 3 different protein domains. (A) Cytochrome b 562, a unmarried-domain protein involved in electron transport in mitochondria. This poly peptide is composed nearly entirely of α helices. (B) The NAD-binding domain of the enzyme lactic (more...)
Few of the Many Possible Polypeptide Chains Will Be Useful
Since each of the 20 amino acids is chemically singled-out and each tin, in principle, occur at whatsoever position in a protein chain, there are twenty × twenty × twenty × twenty = 160,000 unlike possible polypeptide bondage four amino acids long, or 20 n unlike possible polypeptide chains due north amino acids long. For a typical protein length of most 300 amino acids, more than 10390 (20300) different polypeptide chains could theoretically be fabricated. This is such an enormous number that to produce just one molecule of each kind would require many more atoms than exist in the universe.
Simply a very small fraction of this vast set of conceivable polypeptide chains would adopt a single, stable three-dimensional conformation—by some estimates, less than one in a billion. The vast majority of possible protein molecules could adopt many conformations of roughly equal stability, each conformation having different chemical backdrop. And yet almost all proteins nowadays in cells adopt unique and stable conformations. How is this possible? The answer lies in natural selection. A poly peptide with an unpredictably variable structure and biochemical activity is unlikely to help the survival of a cell that contains it. Such proteins would therefore take been eliminated by natural selection through the enormously long trial-and-error process that underlies biological evolution.
Because of natural choice, not only is the amino acrid sequence of a present-24-hour interval poly peptide such that a single conformation is extremely stable, but this conformation has its chemical properties finely tuned to enable the protein to perform a item catalytic or structural function in the cell. Proteins are and so precisely built that the modify of even a few atoms in i amino acid can sometimes disrupt the construction of the whole molecule so severely that all part is lost.
Proteins Tin Be Classified into Many Families
Once a protein had evolved that folded upward into a stable conformation with useful properties, its structure could be modified during evolution to enable it to perform new functions. This process has been profoundly accelerated by genetic mechanisms that occasionally produce indistinguishable copies of genes, allowing one gene copy to evolve independently to perform a new function (discussed in Affiliate seven). This type of event has occurred quite often in the past; every bit a result, many present-twenty-four hour period proteins tin be grouped into protein families, each family member having an amino acrid sequence and a three-dimensional conformation that resemble those of the other family members.
Consider, for example, the serine proteases, a large family of protein-cleaving (proteolytic) enzymes that includes the digestive enzymes chymotrypsin, trypsin, and elastase, and several proteases involved in blood clotting. When the protease portions of any two of these enzymes are compared, parts of their amino acid sequences are found to match. The similarity of their 3-dimensional conformations is even more striking: most of the detailed twists and turns in their polypeptide bondage, which are several hundred amino acids long, are virtually identical (Figure 3-14). The many different serine proteases withal accept distinct enzymatic activities, each cleaving different proteins or the peptide bonds between dissimilar types of amino acids. Each therefore performs a distinct role in an organism.

Figure 3-14
The conformations of two serine proteases compared. The courage conformations of elastase and chymotrypsin. Although merely those amino acids in the polypeptide chain shaded in green are the same in the ii proteins, the 2 conformations are very like (more...)
The story we have told for the serine proteases could be repeated for hundreds of other protein families. In many cases the amino acid sequences have diverged much further than for the serine proteases, then that one cannot be sure of a family relationship between two proteins without determining their three-dimensional structures. The yeast α2 protein and the Drosophila engrailed protein, for example, are both gene regulatory proteins in the homeodomain family. Because they are identical in only 17 of their threescore amino acid residues, their relationship became certain only when their 3-dimensional structures were compared (Figure three-fifteen).

Figure 3-15
A comparing of a class of DNA-binding domains, called homeodomains, in a pair of proteins from 2 organisms separated by more than than a billion years of evolution. (A) A ribbon model of the structure mutual to both proteins. (B) A trace of the α-carbon (more than...)
The various members of a large protein family ofttimes have distinct functions. Some of the amino acid changes that make family unit members unlike were no incertitude selected in the course of evolution because they resulted in useful changes in biological activity, giving the individual family members the unlike functional properties they have today. Simply many other amino acid changes are finer "neutral," having neither a beneficial nor a damaging issue on the bones construction and office of the protein. In addition, since mutation is a random procedure, in that location must as well have been many deleterious changes that altered the three-dimensional structure of these proteins sufficiently to harm them. Such faulty proteins would have been lost whenever the individual organisms making them were at plenty of a disadvantage to be eliminated by natural selection.
Protein families are readily recognized when the genome of any organism is sequenced; for example, the determination of the Dna sequence for the entire genome of the nematode Caenorhabditis elegans has revealed that this tiny worm contains more than eighteen,000 genes. Through sequence comparisons, the products of a large fraction of these genes tin be seen to contain domains from one or another protein family unit; for instance, there appear to exist 388 genes containing protein kinase domains, 66 genes containing DNA and RNA helicase domains, 43 genes containing SH2 domains, 70 genes containing immunoglobulin domains, and 88 genes containing DNA-binding homeodomains in this genome of 97 million base pairs (Figure 3-16).
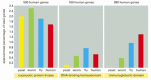
Figure 3-16
Percent of total genes containing one or more copies of the indicated protein domain, as derived from consummate genome sequences. Annotation that ane of the three domains selected, the immunoglobulin domain, has been a relatively belatedly add-on, and its relative (more...)
Proteins Tin can Adopt a Limited Number of Different Protein Folds
It is astounding to consider the rapidity of the increment in our knowledge virtually cells. In 1950, we did non know the order of the amino acids in a single protein, and many fifty-fifty doubted that the amino acids in proteins are bundled in an exact sequence. In 1960, the starting time three-dimensional structure of a protein was determined by x-ray crystallography. Now that we have access to hundreds of thousands of protein sequences from sequencing the genes that encode them, what technical developments tin can we await forrad to next?
It is no longer a big step to progress from a gene sequence to the production of large amounts of the pure protein encoded past that gene. Thanks to DNA cloning and genetic technology techniques (discussed in Chapter 8), this step is often routine. Just there is still zero routine about determining the complete 3-dimensional structure of a protein. The standard technique based on ten-ray diffraction requires that the poly peptide be subjected to conditions that cause the molecules to amass into a large, perfectly ordered crystalline array—that is, a protein crystal. Each protein behaves quite differently in this respect, and poly peptide crystals can be generated only through exhaustive trial-and-error methods that often have many years to succeed—if they succeed at all.
Membrane proteins and big poly peptide complexes with many moving parts take more often than not been the about difficult to crystallize, which is why only a few such protein structures are displayed in this book. Increasingly, therefore, large proteins accept been analyzed through determination of the structures of their individual domains: either by crystallizing isolated domains then bombarding the crystals with 10-rays, or by studying the conformations of isolated domains in concentrated aqueous solutions with powerful nuclear magnetic resonance (NMR) techniques (discussed in Chapter 8). From a combination of x-ray and NMR studies, we at present know the three-dimensional shapes, or conformations, of thousands of different proteins.
By carefully comparison the conformations of known proteins, structural biologists (that is, experts on the structure of biological molecules) have concluded that at that place are a limited number of ways in which protein domains fold upward—mayhap as few as 2000. As we saw, the structures for about 1000 of these protein folds have thus far been determined; we may, therefore, already know half of the total number of possible structures for a protein domain. A consummate catalog of all of the protein folds that be in living organisms would therefore seem to be within our achieve.
Sequence Homology Searches Can Identify Close Relatives
The present database of known protein sequences contains more 500,000 entries, and it is growing very rapidly as more and more genomes are sequenced—revealing huge numbers of new genes that encode proteins. Powerful computer search programs are available that let ane to compare each newly discovered poly peptide with this entire database, looking for possible relatives. Homologous proteins are divers every bit those whose genes have evolved from a common ancestral gene, and these are identified by the discovery of statistically significant similarities in amino acid sequences.
With such a large number of proteins in the database, the search programs discover many nonsignificant matches, resulting in a background noise level that makes information technology very difficult to option out all but the closest relatives. Mostly speaking, a 30% identity in the sequence of two proteins is needed to be certain that a match has been found. However, many short signature sequences ("fingerprints") indicative of particular protein functions are known, and these are widely used to find more afar homologies (Figure iii-17).

Figure iii-17
The use of short signature sequences to observe homologous poly peptide domains. The two brusk sequences of fifteen and ix amino acids shown (green) tin be used to search large databases for a protein domain that is found in many proteins, the SH2 domain. Hither, the (more...)
These poly peptide comparisons are of import because related structures often imply related functions. Many years of experimentation can be saved by discovering that a new poly peptide has an amino acid sequence homology with a protein of known function. Such sequence homologies, for example, showtime indicated that certain genes that crusade mammalian cells to become malignant are protein kinases. In the aforementioned way, many of the proteins that command pattern formation during the embryonic development of the fruit fly Drosophila were chop-chop recognized to exist gene regulatory proteins.
Computational Methods Permit Amino Acrid Sequences to Be Threaded into Known Protein Folds
We know that there are an enormous number of ways to make proteins with the aforementioned three-dimensional structure, and that—over evolutionary time—random mutations can cause amino acid sequences to change without a major change in the conformation of a protein. For this reason, i current goal of structural biologists is to determine all the different poly peptide folds that proteins have in nature, and to devise computer-based methods to test the amino acid sequence of a domain to place which one of these previously determined conformations the domain is likely to adopt.
A computational technique called threading can be used to fit an amino acid sequence to a particular protein fold. For each possible fold known, the figurer searches for the best fit of the particular amino acid sequence to that structure. Are the hydrophobic residues on the within? Are the sequences with a strong propensity to form an α helix in an α helix? And then on. The best fit gets a numerical score reflecting the estimated stability of the structure.
In many cases, one particular three-dimensional construction will stand out every bit a good fit for the amino acid sequence, suggesting an approximate conformation for the protein domain. In other cases, none of the known folds will seem possible. By applying x-ray and NMR studies to the latter class of proteins, structural biologists hope to able to expand the number of known folds chop-chop, aiming for a database that contains the complete library of protein folds that be in nature. With such a library, plus expected improvements in the computational methods used for threading, information technology may eventually become possible to obtain an approximate three-dimensional structure for a protein as before long as its amino acrid sequence is known.
Some Poly peptide Domains, Chosen Modules, Form Parts of Many Different Proteins
As previously stated, about proteins are composed of a series of protein domains, in which different regions of the polypeptide concatenation have folded independently to course compact structures. Such multidomain proteins are believed to have originated when the Dna sequences that encode each domain accidentally became joined, creating a new factor. Novel binding surfaces have often been created at the juxtaposition of domains, and many of the functional sites where proteins bind to pocket-size molecules are establish to exist located there (for an example come across Effigy 3-12). Many large proteins testify clear signs of having evolved by the joining of preexisting domains in new combinations, an evolutionary process called domain shuffling (Figure 3-18).

Figure three-18
Domain shuffling. An extensive shuffling of blocks of protein sequence (protein domains) has occurred during protein evolution. Those portions of a protein denoted by the same shape and colour in this diagram are evolutionarily related. Serine proteases (more...)
A subset of protein domains accept been particularly mobile during evolution; these and then-called protein modules are by and large somewhat smaller (40–200 amino acids) than an average domain, and they seem to have especially versatile structures. The structure of one such module, the SH2 domain, was illustrated in Console three-2 (pp. 138–139). The structures of some boosted protein modules are illustrated in Effigy iii-19.

Figure iii-19
The 3-dimensional structures of some protein modules. In these ribbon diagrams, β-sheet strands are shown as arrows, and the North- and C-termini are indicated by red spheres. (Adapted from One thousand. Baron, D.G. Norman, and I.D. Campbell, Trends Biochem. (more...)
Each of the modules shown has a stable core structure formed from strands of β sheet, from which less-ordered loops of polypeptide chain beetle (greenish). The loops are ideally situated to form binding sites for other molecules, equally most flagrantly demonstrated for the immunoglobulin fold, which forms the basis for antibody molecules (see Effigy 3-42). The evolutionary success of such β-canvass-based modules is likely to have been due to their providing a convenient framework for the generation of new bounden sites for ligands through modest changes to these protruding loops.
A 2nd feature of protein modules that explains their utility is the ease with which they can exist integrated into other proteins. Five of the six modules illustrated in Figure 3-19 accept their N- and C-terminal ends at contrary poles of the module. This "in-line" arrangement ways that when the DNA encoding such a module undergoes tandem duplication, which is non unusual in the development of genomes (discussed in Chapter 7), the duplicated modules can be readily linked in series to class extended structures—either with themselves or with other in-line modules (Figure three-twenty). Strong extended structures equanimous of a series of modules are particularly common in extracellular matrix molecules and in the extracellular portions of jail cell-surface receptor proteins. Other modules, including the SH2 domain and the kringle module illustrated in Figure 3-19, are of a "plug-in" type. After genomic rearrangements, such modules are ordinarily accommodated as an insertion into a loop region of a second protein.

Figure 3-20
An extended construction formed from a serial of in-line protein modules. Four fibronectin blazon 3 modules (run across Figure 3-19) from the extracellular matrix molecule fibronectin are illustrated in (A) ribbon and (B) infinite-filling models. (Adapted from D.J. (more...)
The Human Genome Encodes a Complex Set of Proteins, Revealing Much That Remains Unknown
The result of sequencing the human genome has been surprising, because it reveals that our chromosomes incorporate only 30,000 to 35,000 genes. With regard to gene number, we would appear to be no more than than 1.4-fold more complex than the tiny mustard weed, Arabidopsis, and less than two-fold more complex than a nematode worm. The genome sequences as well reveal that vertebrates have inherited nearly all of their protein domains from invertebrates—with merely 7 percent of identified man domains being vertebrate-specific.
Each of our proteins is on boilerplate more complicated, however. A process of domain shuffling during vertebrate evolution has given rise to many novel combinations of protein domains, with the consequence that in that location are nearly twice every bit many combinations of domains establish in human proteins equally in a worm or a fly. Thus, for example, the trypsinlike serine protease domain is linked to at least xviii other types of poly peptide domains in human being proteins, whereas it is establish covalently joined to just 5 unlike domains in the worm. This extra variety in our proteins greatly increases the range of protein–protein interactions possible (see Figure 3-78), merely how it contributes to making us human is not known.
The complexity of living organisms is staggering, and it is quite sobering to note that we currently lack even the tiniest hint of what the office might be for more 10,000 of the proteins that have thus far been identified in the human being genome. There are certainly enormous challenges ahead for the next generation of cell biologists, with no shortage of fascinating mysteries to solve.
Larger Poly peptide Molecules Often Contain More Than I Polypeptide Chain
The same weak noncovalent bonds that enable a poly peptide chain to fold into a specific conformation also allow proteins to bind to each other to produce larger structures in the cell. Whatever region of a protein's surface that can interact with some other molecule through sets of noncovalent bonds is chosen a binding site. A protein can comprise binding sites for a variety of molecules, both large and modest. If a binding site recognizes the surface of a second protein, the tight binding of two folded polypeptide chains at this site creates a larger protein molecule with a precisely divers geometry. Each polypeptide chain in such a protein is called a protein subunit.
In the simplest case, ii identical folded polypeptide chains bind to each other in a "head-to-head" organisation, forming a symmetric complex of two poly peptide subunits (a dimer) held together by interactions between two identical binding sites. The Cro repressor protein—a gene regulatory protein that binds to Dna to plow genes off in a bacterial cell—provides an case (Effigy 3-21). Many other types of symmetric poly peptide complexes, formed from multiple copies of a single polypeptide concatenation, are normally found in cells. The enzyme neuraminidase, for example, consists of four identical poly peptide subunits, each jump to the adjacent in a "head-to-tail" arrangement that forms a closed band (Figure 3-22).

Figure 3-21
2 identical protein subunits bounden together to form a symmetric protein dimer. The Cro repressor protein from bacteriophage lambda binds to DNA to plough off viral genes. Its two identical subunits bind head-to-head, held together by a combination of (more...)

Figure 3-22
A protein molecule containing multiple copies of a unmarried protein subunit. The enzyme neuraminidase exists as a ring of 4 identical polypeptide chains. The small diagram shows how the repeated use of the same binding interaction forms the construction. (more...)
Many of the proteins in cells comprise two or more types of polypeptide chains. Hemoglobin, the protein that carries oxygen in red claret cells, is a especially well-studied example (Figure 3-23). It contains 2 identical α-globin subunits and two identical β-globin subunits, symmetrically arranged. Such multisubunit proteins are very mutual in cells, and they can be very big. Figure 3-24 provides a sampling of proteins whose exact structures are known, allowing the sizes and shapes of a few larger proteins to be compared with the relatively small proteins that we have thus far presented as models.
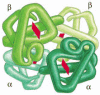
Figure 3-23
A protein formed equally a symmetric assembly of two different subunits. Hemoglobin is an abundant protein in ruby-red claret cells that contains 2 copies of α globin and 2 copies of β globin. Each of these four polypeptide chains contains a (more...)

Figure 3-24
A collection of protein molecules, shown at the same scale. For comparison, a DNA molecule bound to a protein is as well illustrated. These space-filling models stand for a range of sizes and shapes. Hemoglobin, catalase, porin, alcohol dehydrogenase, and (more...)
Some Proteins Form Long Helical Filaments
Some protein molecules can gather to course filaments that may bridge the entire length of a cell. Nigh simply, a long chain of identical protein molecules can be constructed if each molecule has a binding site complementary to some other region of the surface of the same molecule (Figure 3-25). An actin filament, for example, is a long helical structure produced from many molecules of the protein actin (Effigy 3-26). Actin is very arable in eucaryotic cells, where it constitutes one of the major filament systems of the cytoskeleton (discussed in Affiliate 16).
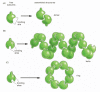
Effigy iii-25
Protein assemblies. (A) A protein with but 1 binding site can form a dimer with another identical protein. (B) Identical proteins with two unlike bounden sites frequently form a long helical filament. (C) If the two binding sites are tending appropriately (more...)

Figure iii-26
Actin filaments. (A) Transmission electron micrographs of negatively stained actin filaments. (B) The helical organisation of actin molecules in an actin filament. (A, courtesy of Roger Craig.)
Why is a helix such a common structure in biology? As we have seen, biological structures are frequently formed by linking subunits that are very like to each other—such every bit amino acids or protein molecules—into long, repetitive chains. If all the subunits are identical, the neighboring subunits in the chain tin often fit together in merely 1 way, adjusting their relative positions to minimize the complimentary energy of the contact between them. As a result, each subunit is positioned in exactly the aforementioned way in relation to the side by side, and so that subunit 3 fits onto subunit 2 in the aforementioned way that subunit 2 fits onto subunit 1, and and then on. Considering information technology is very rare for subunits to join upwardly in a directly line, this arrangement mostly results in a helix—a regular structure that resembles a screw staircase, as illustrated in Figure 3-27. Depending on the twist of the staircase, a helix is said to be either right-handed or left-handed (Figure 3-27E). Handedness is not affected by turning the helix upside downwardly, but it is reversed if the helix is reflected in the mirror.

Figure 3-27
Some properties of a helix. (A–D) A helix forms when a series of subunits demark to each other in a regular style. At the bottom, the interaction between ii subunits is shown; behind them are the helices that result. These helices accept ii (A), three (more...)
Helices occur commonly in biological structures, whether the subunits are small-scale molecules linked together by covalent bonds (for case, the amino acids in an α helix) or large protein molecules that are linked past noncovalent forces (for example, the actin molecules in actin filaments). This is not surprising. A helix is an unexceptional structure, and information technology is generated simply by placing many similar subunits next to each other, each in the aforementioned strictly repeated relationship to the i before.
A Protein Molecule Can Accept an Elongated, Gristly Shape
Most of the proteins we take discussed so far are globular proteins, in which the polypeptide concatenation folds upwardly into a compact shape like a ball with an irregular surface. Enzymes tend to be globular proteins: even though many are big and complicated, with multiple subunits, most have an overall rounded shape (see Figure iii-24). In contrast, other proteins take roles in the cell requiring each private protein molecule to bridge a large altitude. These proteins generally take a relatively simple, elongated three-dimensional construction and are usually referred to as gristly proteins.
Ane large family of intracellular fibrous proteins consists of α-keratin, introduced earlier, and its relatives. Keratin filaments are extremely stable and are the main component in long-lived structures such every bit hair, horn, and nails. An α-keratin molecule is a dimer of 2 identical subunits, with the long α helices of each subunit forming a coiled-roll (run into Figure three-eleven). The coiled-whorl regions are capped at each end by globular domains containing binding sites. This enables this class of poly peptide to assemble into ropelike intermediate filaments—an important component of the cytoskeleton that creates the jail cell's internal structural scaffold (see Figure sixteen-16).
Fibrous proteins are especially abundant outside the cell, where they are a primary component of the gel-like extracellular matrix that helps to demark collections of cells together to form tissues. Extracellular matrix proteins are secreted by the cells into their surroundings, where they frequently get together into sheets or long fibrils. Collagen is the most arable of these proteins in brute tissues. A collagen molecule consists of three long polypeptide chains, each containing the nonpolar amino acid glycine at every third position. This regular structure allows the bondage to wind effectually i another to generate a long regular triple helix (Figure 3-28A). Many collagen molecules then demark to one another side-past-side and end-to-terminate to create long overlapping arrays—thereby generating the extremely tough collagen fibrils that give connective tissues their tensile strength, as described in Chapter 19.

Figure 3-28
Collagen and elastin. (A) Collagen is a triple helix formed by three extended protein chains that wrap around one another (bottom). Many rodlike collagen molecules are cantankerous-linked together in the extracellular infinite to form unextendable collagen fibrils (more...)
In complete contrast to collagen is another protein in the extracellular matrix, elastin. Elastin molecules are formed from relatively loose and unstructured polypeptide chains that are covalently cantankerous-linked into a rubberlike elastic meshwork: unlike most proteins, they exercise non take a uniquely defined stable structure, merely tin be reversibly pulled from i conformation to another, as illustrated in Figure iii-28B. The resulting elastic fibers enable skin and other tissues, such equally arteries and lungs, to stretch and recoil without trigger-happy.
Extracellular Proteins Are Often Stabilized past Covalent Cantankerous-Linkages
Many protein molecules are either attached to the outside of a cell'due south plasma membrane or secreted every bit office of the extracellular matrix. All such proteins are straight exposed to extracellular weather condition. To help maintain their structures, the polypeptide chains in such proteins are often stabilized by covalent cross-linkages. These linkages can either necktie two amino acids in the same protein together, or connect different polypeptide bondage in a multisubunit protein. The nigh common cross-linkages in proteins are covalent sulfur–sulfur bonds. These disulfide bonds (too called S–S bonds) grade as proteins are being prepared for consign from cells. As described in Chapter 12, their formation is catalyzed in the endoplasmic reticulum by an enzyme that links together 2 pairs of –SH groups of cysteine side chains that are adjacent in the folded protein (Figure 3-29). Disulfide bonds do not change the conformation of a poly peptide only instead human action every bit atomic staples to reinforce its most favored conformation. For case, lysozyme—an enzyme in tears that dissolves bacterial cell walls—retains its antibacterial activity for a long time because it is stabilized by such cross-linkages.
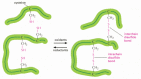
Figure 3-29
Disulfide bonds. This diagram illustrates how covalent disulfide bonds form betwixt adjacent cysteine side chains. Equally indicated, these cantankerous-linkages tin bring together either two parts of the same polypeptide concatenation or two unlike polypeptide chains. Since the (more...)
Disulfide bonds by and large fail to form in the cell cytosol, where a high concentration of reducing agents converts S–Due south bonds dorsum to cysteine –SH groups. Apparently, proteins practice non require this type of reinforcement in the relatively mild environment inside the cell.
Poly peptide Molecules Often Serve as Subunits for the Assembly of Large Structures
The same principles that enable a protein molecule to associate with itself to grade rings or filaments operate to generate much larger structures in the cell—supramolecular structures such equally enzyme complexes, ribosomes, protein filaments, viruses, and membranes. These large objects are not fabricated as single, behemothic, covalently linked molecules. Instead they are formed by the noncovalent associates of many separately manufactured molecules, which serve as the subunits of the last structure.
The use of smaller subunits to build larger structures has several advantages:
- i.
-
A large structure built from one or a few repeating smaller subunits requires only a small corporeality of genetic data.
- ii.
-
Both associates and disassembly tin exist readily controlled, reversible processes, since the subunits associate through multiple bonds of relatively low energy.
- three.
-
Errors in the synthesis of the structure can be more than easily avoided, since correction mechanisms can operate during the course of assembly to exclude malformed subunits.
Some poly peptide subunits assemble into flat sheets in which the subunits are arranged in hexagonal patterns. Specialized membrane proteins are sometimes bundled this manner in lipid bilayers. With a slight change in the geometry of the individual subunits, a hexagonal sheet can be converted into a tube (Figure three-30) or, with more changes, into a hollow sphere. Protein tubes and spheres that bind specific RNA and Dna molecules form the coats of viruses.

Effigy three-30
An example of single protein subunit assembly requiring multiple poly peptide–protein contacts. Hexagonally packed globular protein subunits can course either a apartment sheet or a tube.
The formation of airtight structures, such as rings, tubes, or spheres, provides additional stability considering it increases the number of bonds between the poly peptide subunits. Moreover, because such a construction is created by mutually dependent, cooperative interactions between subunits, information technology can be driven to assemble or disassemble by a relatively small alter that affects each subunit individually. These principles are dramatically illustrated in the protein coat or capsid of many simple viruses, which takes the form of a hollow sphere (Effigy 3-31). Capsids are oft made of hundreds of identical poly peptide subunits that enclose and protect the viral nucleic acid (Figure 3-32). The protein in such a capsid must have a peculiarly adjustable structure: it must not only make several different kinds of contacts to create the sphere, it must also alter this arrangement to let the nucleic acid out to initiate viral replication in one case the virus has entered a cell.

Figure three-31
The capsids of some viruses, all shown at the same calibration. (A) Tomato bushy stunt virus; (B) poliovirus; (C) simian virus forty (SV40); (D) satellite tobacco necrosis virus. The structures of all of these capsids take been determined by x-ray crystallography (more...)

Figure 3-32
The construction of a spherical virus. In many viruses, identical protein subunits pack together to create a spherical beat out (a capsid) that encloses the viral genome, composed of either RNA or Deoxyribonucleic acid (see also Figure 3-31). For geometric reasons, no more than (more...)
Many Structures in Cells Are Capable of Cocky-Assembly
The information for forming many of the complex assemblies of macromolecules in cells must exist contained in the subunits themselves, considering purified subunits can spontaneously gather into the final structure under the appropriate atmospheric condition. The first large macromolecular amass shown to exist capable of self-assembly from its component parts was tobacco mosaic virus (TMV). This virus is a long rod in which a cylinder of poly peptide is arranged around a helical RNA core (Figure three-33). If the dissociated RNA and protein subunits are mixed together in solution, they recombine to form fully agile viral particles. The assembly procedure is unexpectedly complex and includes the formation of double rings of protein, which serve as intermediates that add to the growing viral coat.

Figure 3-33
The structure of tobacco mosaic virus (TMV). (A) An electron micrograph of the viral particle, which consists of a single long RNA molecule enclosed in a cylindrical protein coat composed of identical protein subunits. (B) A model showing part of the (more than...)
Some other complex macromolecular aggregate that can reassemble from its component parts is the bacterial ribosome. This structure is composed of about 55 unlike protein molecules and 3 different rRNA molecules. If the individual components are incubated nether appropriate conditions in a test tube, they spontaneously re-class the original structure. Most importantly, such reconstituted ribosomes are able to perform protein synthesis. As might exist expected, the reassembly of ribosomes follows a specific pathway: after certain proteins have bound to the RNA, this complex is then recognized by other proteins, and and so on, until the structure is complete.
It is still non clear how some of the more elaborate self-assembly processes are regulated. Many structures in the prison cell, for case, seem to have a precisely defined length that is many times greater than that of their component macromolecules. How such length determination is accomplished is in many cases a mystery. Three possible mechanisms are illustrated in Figure 3-34. In the simplest example, a long cadre protein or other macromolecule provides a scaffold that determines the extent of the final assembly. This is the machinery that determines the length of the TMV particle, where the RNA concatenation provides the core. Similarly, a core protein is thought to determine the length of the thin filaments in muscle, as well as the length of the long tails of some bacterial viruses (Figure 3-35).

Figure 3-34
Three mechanisms of length determination for large protein assemblies. (A) Coassembly along an elongated core protein or other macromolecule that acts as a measuring device. (B) Termination of assembly because of strain that accumulates in the polymeric (more...)
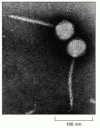
Figure iii-35
An electron micrograph of bacteriophage lambda. The tip of the virus tail attaches to a specific poly peptide on the surface of a bacterial cell, afterward which the tightly packaged Deoxyribonucleic acid in the head is injected through the tail into the cell. The tail has a precise (more than...)
The Formation of Complex Biological Structures Is Often Aided by Assembly Factors
Not all cellular structures held together past noncovalent bonds are capable of cocky-associates. A mitochondrion, a cilium, or a myofibril of a muscle jail cell, for example, cannot form spontaneously from a solution of its component macromolecules. In these cases, part of the assembly information is provided by special enzymes and other cellular proteins that perform the function of templates, guiding structure simply taking no role in the last assembled structure.
Even relatively simple structures may lack some of the ingredients necessary for their ain associates. In the formation of certain bacterial viruses, for example, the head, which is composed of many copies of a single protein subunit, is assembled on a temporary scaffold composed of a second poly peptide. Because the 2d protein is absent from the final viral particle, the caput construction cannot spontaneously reassemble once it has been taken apart. Other examples are known in which proteolytic cleavage is an essential and irreversible footstep in the normal assembly procedure. This is even the case for some small poly peptide assemblies, including the structural protein collagen and the hormone insulin (Figure three-36). From these relatively uncomplicated examples, information technology seems very likely that the associates of a structure as complex as a mitochondrion or a cilium volition involve temporal and spatial ordering imparted by numerous other cell components.
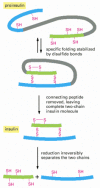
Figure 3-36
Proteolytic cleavage in insulin assembly. The polypeptide hormone insulin cannot spontaneously re-form efficiently if its disulfide bonds are disrupted. It is synthesized as a larger protein (proinsulin) that is broken by a proteolytic enzyme after the (more...)
Summary
The 3-dimensional conformation of a protein molecule is determined by its amino acid sequence. The folded construction is stabilized by noncovalent interactions between unlike parts of the polypeptide chain. The amino acids with hydrophobic side chains tend to cluster in the interior of the molecule, and local hydrogen-bond interactions between neighboring peptide bonds give rise to α helices and β sheets.
Globular regions, known equally domains, are the modular units from which many proteins are constructed; such domains generally contain 40–350 amino acids. Modest proteins typically consist of only a single domain, while large proteins are formed from several domains linked together by curt lengths of polypeptide concatenation. Every bit proteins have evolved, domains have been modified and combined with other domains to construct new proteins. Domains that participate in the formation of large numbers of proteins are known equally protein modules. Thus far, well-nigh 1000 different means of folding up a domain accept been observed, among more than nearly 10,000 known poly peptide structures.
Proteins are brought together into larger structures by the aforementioned noncovalent forces that determine protein folding. Proteins with bounden sites for their own surface tin can assemble into dimers, closed rings, spherical shells, or helical polymers. Although mixtures of proteins and nucleic acids can get together spontaneously into circuitous structures in a test tube, many biological assembly processes involve irreversible steps. Consequently, non all structures in the jail cell are capable of spontaneous reassembly after they have been dissociated into their component parts.

What Atom Helps Makeup Amino Acids, The Disulfide Bonds Help Add Structure To Big Proteins,
Source: https://www.ncbi.nlm.nih.gov/books/NBK26830/
Posted by: bortonfortaryto.blogspot.com
0 Response to "What Atom Helps Makeup Amino Acids, The Disulfide Bonds Help Add Structure To Big Proteins"
Post a Comment